Faron plans to approach the FDA regarding the Phase 2b results, and as we just heard, CR (Complete Response) and its duration are what they are aiming for. One study on the subject; from the portal, click Julie under Zeidan’s Bex presentation—it’s part of that ASH package that caused the “Tänka På” (Food for Thought).
https://www.vjhemonc.com/event/post-ash-2025-mds-highlights/
”Durability of complete response outperforms complete response rates as a surrogate endpoint for advancing to phase III trials in high-risk myelodysplastic syndromes”
Julie Braish University of Texas MD Anderson Cancer Center, Houston, TX, United States
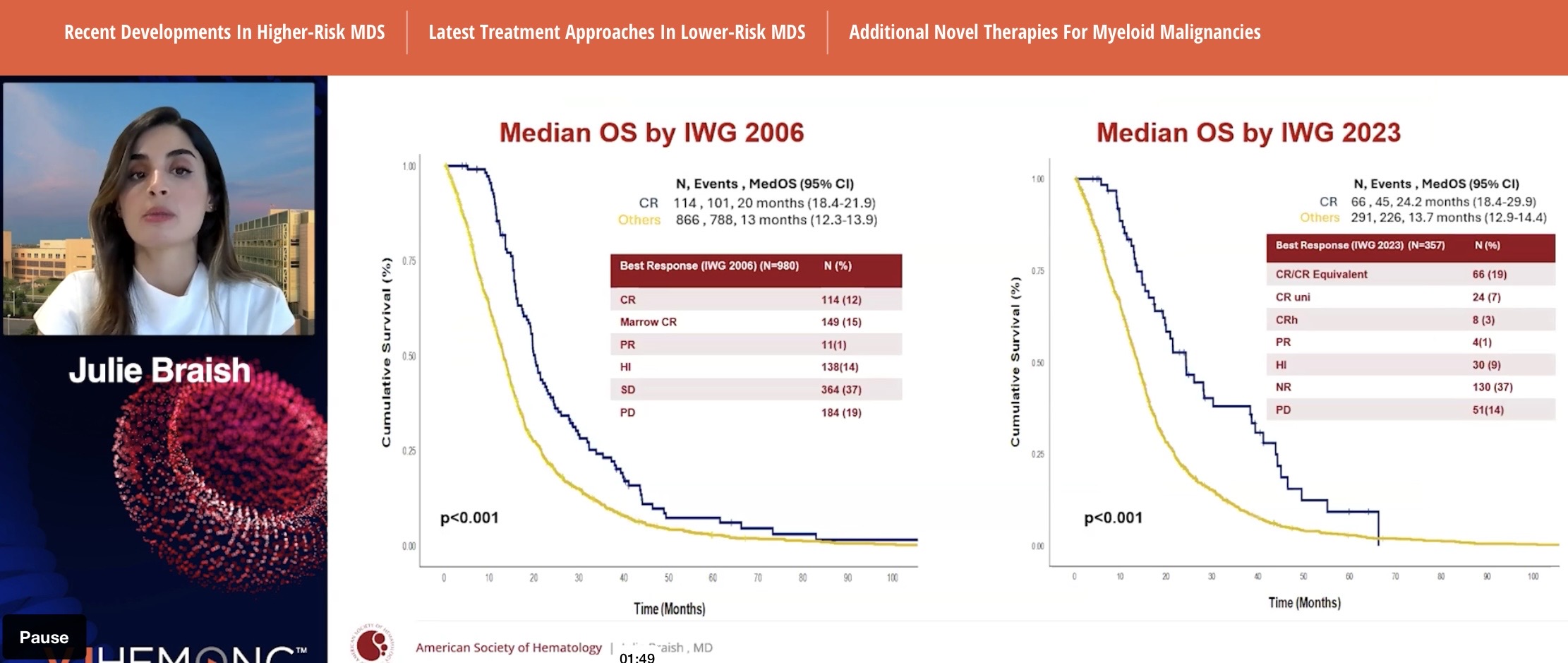
A study conducted at the Moffitt Cancer Center, retrospective (data collected after treatment), 980 HR-MDS patients (intermediate and higher risk).
An investigation into why success in Phase 1/2 does not translate to Phase 3, which is the “hottest” topic right now, as we know… TP53 is particularly problematic. CR is seen there, but it’s short-lived.
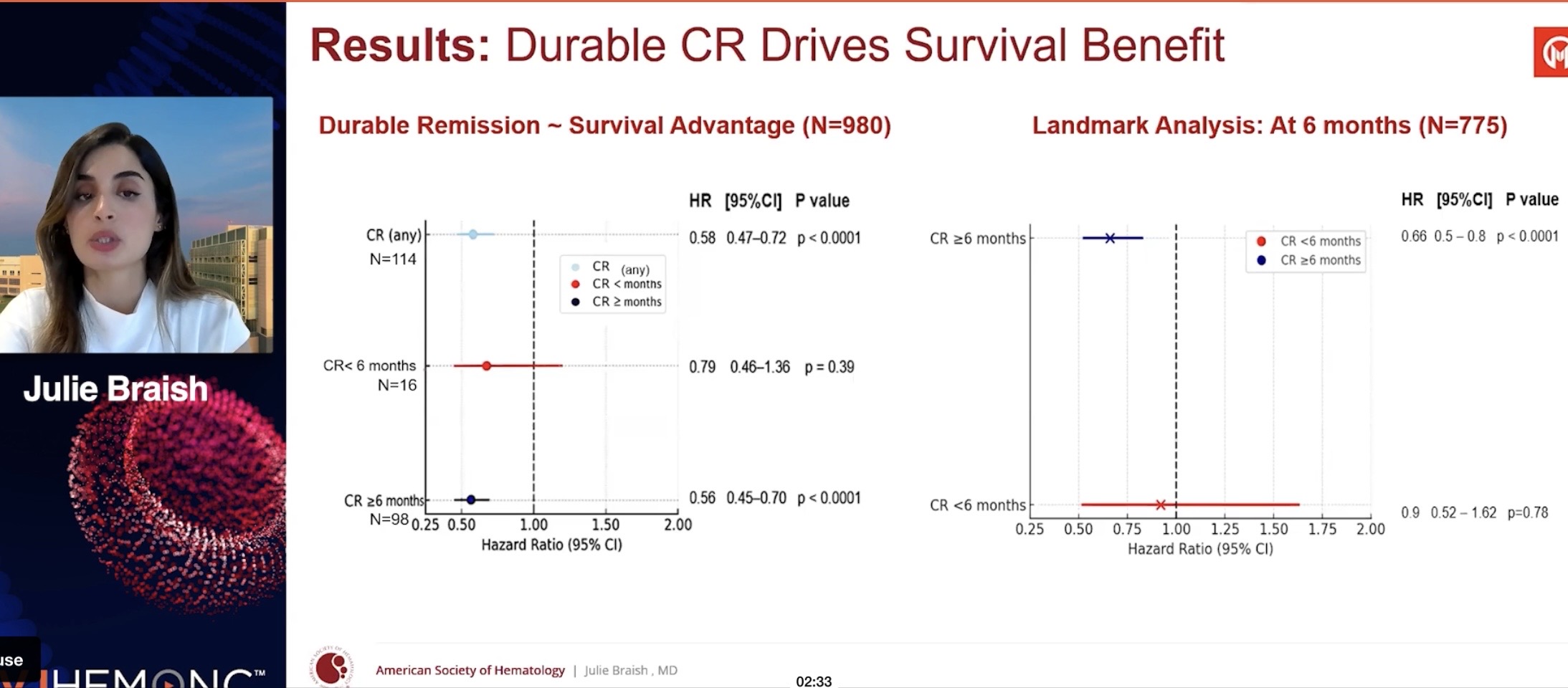
Hypothesis: CR with a duration of 6 months or more predicts survival benefit (OS) better than a one-time CR response.
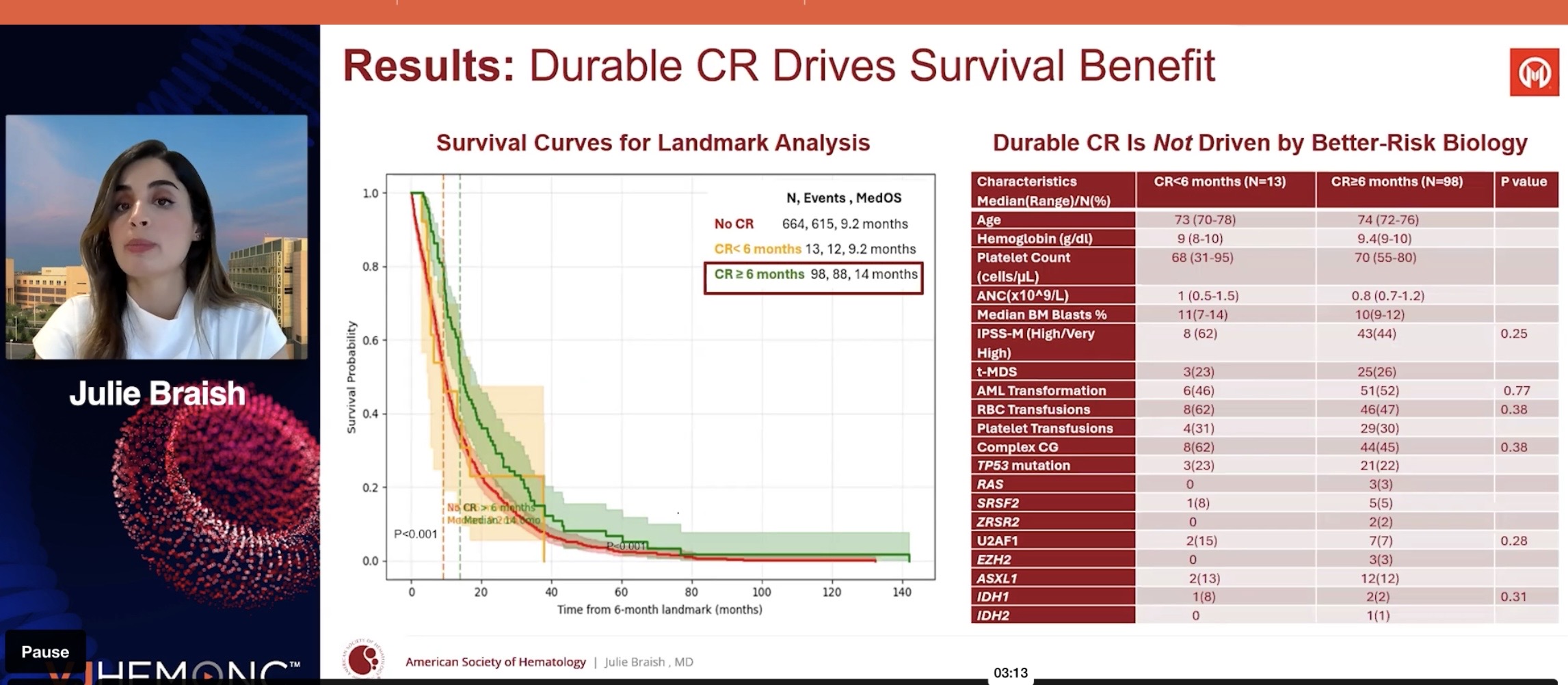
Those undergoing bone marrow transplants were not included in the analysis. (Transplants must be handled in the plan so that they don’t penalize Bex if the CR duration is cut short by a desired transplant, ed. note.)
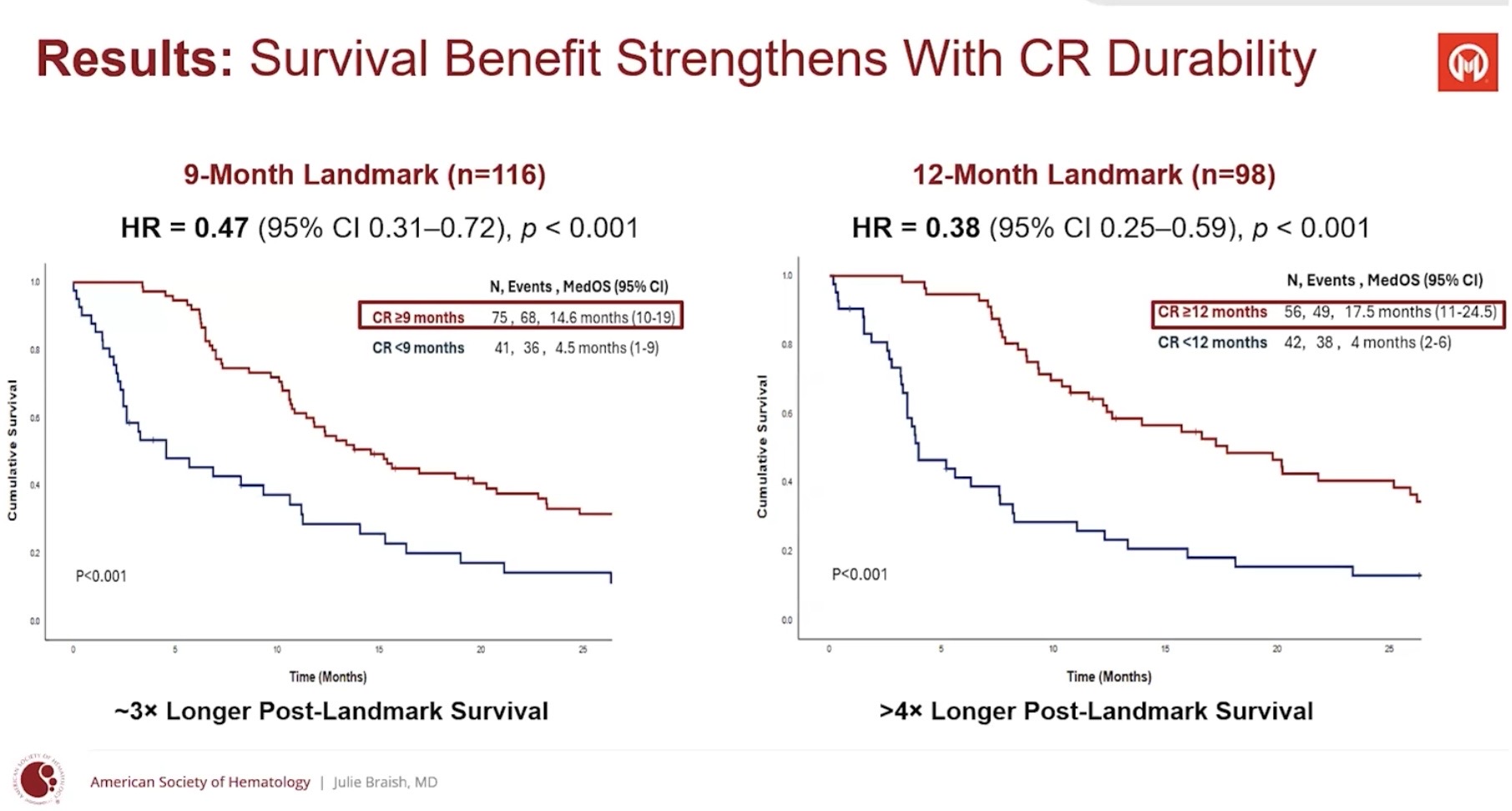
They showed that the longer the CR, the longer the survival (pretty obvious). CR itself also works. In their opinion, CR duration of 6 months or more could be considered a surrogate—an indicator predicting survival benefit (the kind that, if approved by the FDA, could lead to AA [Accelerated Approval] marketing authorization, ed. note). CR has already been such an endpoint. What will be enough for the FDA next year is not known; the guidance only uses the word ”meaningful”.
What does Faron say about those response durations?
business update | February 11, 2026 Faron Pharmaceuticals - Innovatiivisia lääketieteen ratkaisuja (Osa 2) - #3416 käyttäjältä Vino_Pino :
”Follow-up is still immature (as I understand, it was the November reading, ed. note), but the median duration of CR is already over 12 months, and follow-up continues – potentially it will still rise. In TP53-mutated patients, the CR rate was an amazing 70 percent, and the duration of CR was over 10 months, possibly still rising. Historically, this patient group usually lives 8–10 months.”
Looking at those figures, it seems we are starting to be in the safe zone. New numbers likely in a month at BSH. A median of over 12 months means that for at least half of the patients, responses have lasted over 12 months. In the Moffitt study, the 6-month limit is instead the minimum response duration that everyone should exceed.
It could be that “easy” patients are “always” selected for Phase 1/2a, and the truth and reality only emerge in randomized trials. Now it might be the other way around, if/when TP53 patients have been selected more than their natural proportion would be. If preliminary observations that cytopenias occur less with Bex than without hold true, the problems experienced in Phase 3 may partly turn to Bex’s advantage, as there are no dangerous deficiencies like with many other toxic drugs. Well, Phase 2b/3 will eventually show how it goes.
At least “how many cycles” of aza (azacitidine) were received will also be controlled as a factor. Indications of future Phase 2b success can be obtained if these CR-duration surrogates are scrutinized.
Studying R/R (relapsed/refractory) patients finally opened a completely new path forward, the continuation of City of Hope, the IIT (Investigator-Initiated Trial)—we’ll see what kind of support it needs from Phase 2b or what they think about communication with the regulator there